
[Seminar Blog] Reaching maturity? Malaria parasites’ developmental journey through the mosquito11/4/2021
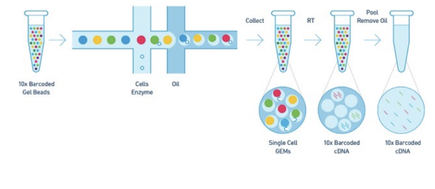
Written by PhD students Arielle Arsenault-Benoit & Minh Le Malaria is a mosquito-borne illness that has plagued human civilization for millennia (Cox 2010), and continues to infect many people, with over 200 million documented cases in 2019 (World Health Organization, 2020). Preventative interventions and treatments are available for those that can access them, yet according to the WHO, there are 400,000 annual deaths worldwide, and nearly 70% of those are children under 5 years old. Among those interventions are insecticide treated bed nets, residual insecticide spraying, preventative drugs, and treatment with combined drug therapy. Effective prevention and treatment efforts, plus advances in diagnostic testing, have greatly reduced both case numbers and mortality rates, but the development of resistance, including insecticide resistance in the mosquito vector population and drug resistance in the pathogen population present new challenges. A greater understanding of the underlying biology of the pathogen may allow for improved and more targeted interventions in the future. At our recent Department of Entomology colloquium, Dr. Haikel Bogale, a Post-Doctoral Fellow at the Institute for Genome Sciences, University of Maryland School of Medicine, discussed his work on the role of differential gene expression on the maturity of malaria parasites at different time points and at different locations within the mosquito body. Malaria is caused by several species of unicellular parasites in the genus Plasmodium. These parasites have a complex life cycle, in which they undergo development, multiplication, and reproduction within the mammalian host and the mosquito vector. A female mosquito picks up the parasite from an infected host’s blood (i.e. human), then, while also going through its developmental stages, the parasite must enter the mosquito midgut via the blood meal, escape the mosquito’s immune system, break out of the midgut and into the body cavity, and travel through the hemolymph – analogous to blood in insects– to the mosquito’s salivary glands. Once in the salivary glands, the infectious stage of Plasmodium, called sporozoite, is transmitted to a mammalian host to cause disease and perpetuate the cycle. In an infected mosquito, thousands of sporozoites reach the salivary glands, but only a handful of those are injected to the mammalian skin. (For more detailed information about the life cycle of Plasmodium parasites, visit the CDC Malaria website.) During an organism’s life cycle, different genes are expressed at different times, locations, and quantities in order to perform vital functions. In other words, individual genes are like different tools that an organism can retrieve and use to address different environmental pressures that they experience throughout their lifespan. The same concept holds true for malaria-causing Plasmodium parasites. Dr. Bogale leverages this notion to study how gene expression of Plasmodium parasites differs between multiple species’ sporozoites and between different anatomical locations in the mosquito where sporozoites reside. When a gene is expressed, a class of molecules known as RNA is produced with a unique sequence. Reading and analyzing the RNA sequence profile of the sporozoites at certain stages can give us an idea of which genes are expressed at a particular stage and how much of that gene is being expressed. The traditional technique used to read the sporozoite RNA sequence is known as bulk RNA sequencing, which generates the RNA profile of a population of sporozoite cells. This technique, however, only produces a big picture of the population’s RNA profile and masks the minor variations between each sporozoite cell in that population (Chen et al., 2019). Dr. Bogale was among the first to employ an alternative technique called single cell RNA sequencing (scRNA-seq) to study Plasmodium sporozoites collected from multiple anatomical sites in the mosquito. As its name suggests, scRNA-seq makes it possible to sequence an individual sporozoite’s RNA profile within a sample containing thousands and thousands of sporozoites. This technique works by separating cells in individual oil droplets and allowing for the capture of information from messenger RNA (mRNA) of each cell (Figure 1), ultimately highlighting the differences in gene expression among all sporozoite cells within a given sample, a feat that is not possible with bulk RNA sequencing.
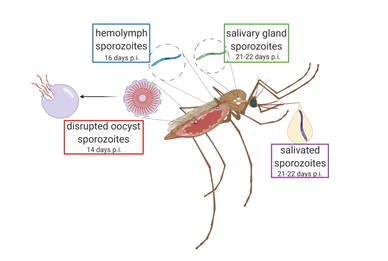
Secondly, Dr. Bogale’s group used scRNA-seq on sporozoites of a single species at four snapshots of their lifespan inside of the mosquito; from when they are just released from the oocyst, to when they are traveling in the mosquito’s hemolymph, when they inhabit the mosquito’s salivary glands, and then finally after they have been expelled from the mosquito’s proboscis by forced salivation (Figure 2). This allowed researchers to determine whether sporozoites within the same area of the mosquito are in the same stage of development and express the same genes. Researchers found RNA profiles from sporozoites that were collected from different mosquito anatomical sites can be similar (e.g., sporozoites from oocysts vs salivary glands), suggesting sporozoite maturation is not solely dependent on mosquito environment. However, they also identified over 50 differentially expressed genes across different sporozoite’s developmental stages (correlating with different anatomical residence). Lastly, they report an upregulation of components necessary for producing protein (ribosomal RNA) after salivation of sporozoites by the mosquito, in contrast to when it resides in the salivary gland. This could signify the transition from the sporozoite’s quiescence phase, where the parasite “sleeps” inside of the mosquito’s salivary gland, to a more active stage when it is trying to assemble the machinery needed to invade mammalian host cells.
References:
Bogale, H.N., Pascini, T.V., Kanatani, S., Sá, J.M., Wellems, T.E., Sinnis, P., Vega-Rodríguez, J. and Serre, D., 2021. Transcriptional heterogeneity and tightly regulated changes in gene expression during Plasmodium berghei sporozoite development. Proceedings of the National Academy of Sciences, 118(10). Chen, G., Ning B., Shi, T., 2019. Single-cell RNA-seq technologies and related computational data analysis. Frontiers in Genetics, 10: 317. Cox, F.E., 2010. History of the discovery of the malaria parasites and their vectors. Parasites Vectors 3(5). World Health Organization, 2020. World malaria report 2020: 20 years of global progress and challenges. In World malaria report 2020: 20 years of global progress and challenges. Authors: Arielle Arsenault-Benoit is a PhD student in the Fritz Lab. She studies the community composition and landscape genomics of Culex mosquitoes, vectors of West Nile virus, across a heterogeneous landscape. You can find her on twitter @BenoitArielle. Minh Le is a PhD student in the Pick Lab. He studies the genetic networks involved in insect segmentation, specifically on hemipteran insects. Comments are closed.
|
Categories
All
Archives
June 2024
|
 RSS Feed
RSS Feed



